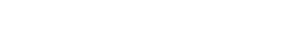






PUNTOS CLAVE
- El cáncer colorrectal (CCR) es una enfermedad de gran magnitud, que ocasiona una importante mortalidad. Las tasas de incidencia se han incrementado de forma significativa en la última década, de tal manera que representa la segunda causa de incidencia y mortalidad por cáncer, tanto en hombres como en mujeres en la mayoría de los países desarrollados, y el primer lugar si consideramos conjuntamente ambos sexos.
- Son factores de riesgo para el CCR una dieta rica en grasas animales, el consumo de alcohol y tabaco, la obesidad, la diabetes mellitus tipo 2, los adenomas colónicos, los antecedentes familiares de esta neoplasia (cáncer colorrectal no asociado a poliposis, poliposis adenomatosa familiar, fundamentalmente), y la enfermedad inflamatoria intestinal.
- La clínica se caracteriza por la presencia de cambios en el ritmo intestinal, rectorragia o hematoquecia, dolor abdominal o, de modo menos frecuente, síndrome constitucional sin focalización digestiva, complicaciones como obstrucción, perforación o la presencia de metástasis a distancia.
- La clave para la disminución de la mortalidad por este cáncer se encuentra en el diagnóstico y el tratamiento temprano de la enfermedad.
- Las distintas sociedades científicas aceptan como técnicas de cribado la prueba de sangre oculta en heces, la sigmoidoscopia y la colonoscopia.
- La colonoscopia es el método más sensible y específico para la detección de pólipos y de cáncer colorrectal. Es la prueba predilecta para la posterior evaluación de los pacientes cuya prueba de sangre oculta en heces ha sido positiva.
- El desarrollo de nuevos equipos endoscópicos, de alta resolución y de amplificación, con la aplicación de técnicas de tinción, posibilita la detección de lesiones diminutas y de pólipos superficiales.
- Existe evidencia de que el cribado del cáncer colorrectal en población de riesgo medio (individuos de edad ≥ 50 años sin otros factores de riesgo para desarrollar cáncer colorrectal), con prueba de sangre oculta en heces anual o bienal, sigmoidoscopia cada 5 años o colonoscopia cada 10 años, disminuye la incidencia y la mortalidad por esta neoplasia.
- Resulta fundamental identificar y hacer un cribado en la población de riesgo elevado (individuos con poliposis adenomatosa familiar, cáncer colorrectal hereditario no poliposis, cáncer colorrectal familiar, antecedentes personales de adenomas colónicos o enfermedad inflamatoria intestinal).
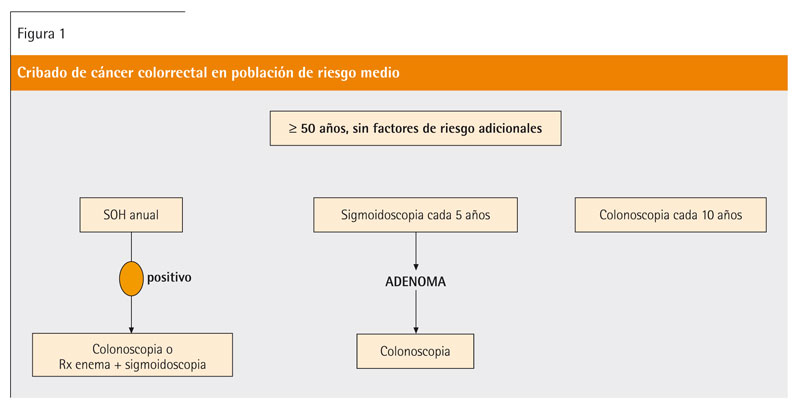
- El riesgo de presentar un cáncer colorrectal familiar está condicionado por el número de familiares afectados, el grado de parentesco y la edad de diagnóstico del cáncer colorrectal en el familiar afectado.
- En España, la mayoría de los planes de salud de las comunidades autónomas establecen como objetivo el cribado del CCR y hacen referencia a la necesidad de realizar estudios piloto que evalúen la viabilidad de la puesta en marcha de programas de cobertura poblacional. Cuatro comunidades autónomas desarrollan actualmente programas de cobertura poblacional (Murcia, Cataluña, Canarias y Valencia).
CRIBADO DEL CCR. PRUEBAS DE CRIBADO. PERIODICIDAD SEGÚN LOS FACTORES DE RIESGO
El cáncer colorrectal (CCR) es una enfermedad de gran magnitud, que ocasiona una importante mortalidad. Las tasas de incidencia se han incrementado de forma significativa en la última década, de tal manera que el CCR representa la segunda causa de incidencia y mortalidad por cáncer, tanto en hombres como en mujeres en la mayoría de los países desarrollados, y el primer lugar si consideramos conjuntamente ambos sexos.
En España, se estima que en el año 2006 se diagnosticaron 14.564 casos de cáncer colorrectal en hombres y 7.766 casos en mujeres, lo que representa una tasa de incidencia ajustada por población europea de 54,4 en hombres y de 25,4 en mujeres, siendo estas tasas más elevadas a partir de los 40 años y, sobre todo, a partir de los 50 años1,2.
El CCR es una patología que, por su elevada prevalencia, incidencia y morbimortalidad, se puede beneficiar de un cribado poblacional (grado de recomendación A)5. La efectividad del cribado se mide en años de vida ganados ajustados a calidad de vida (AVAC) al prevenir el CCR y aumentar las cifras de supervivencia. El cribado del CCR, incluso en el caso de que el cumplimiento sea imperfecto, reduce significativamente la mortalidad por este tumor y los costes son inferiores que los debidos al cribado de otras patologías (cáncer de mama o cérvix) en cuanto a AVAC (grado de recomendación A)4,5.
El objetivo del cribado es la detección de la presencia de lesiones precancerosas (adenomas) o de cáncer en individuos asintomáticos, permitiendo así el tratamiento precoz y el aumento de la supervivencia. El CCR tiene una lesión precursora, el pólipo adenomatoso, de lento crecimiento y fácilmente identificable, sobre el que se puede actuar mediante polipectomía. El período desde la primera aparición de un pólipo hasta el de- sarrollo de cáncer oscila, probablemente, entre 5 y 15 años3,5,6.
Hay que clasificar el nivel de riesgo individual de padecer CCR para determinar cuándo debe iniciarse el programa de cribado, a quién debe ofrecérsele, qué tipo de pruebas es preciso realizar y con qué frecuencia se deben llevar a cabo. Por tanto, la primera consideración que se debe tener en cuenta en el cribado de esta patología es determinar si el paciente presenta un riesgo bajo, medio o elevado. Aunque el CCR es más probable en individuos con riesgo moderado o alto, a escala poblacional la mayoría de los casos inciden en personas con riesgo medio. Para la valoración del riesgo de un individuo en relación con el desarrollo de esta patología es fundamental la evaluación de los antecedentes personales y/o familiares. Se debe realizar una correcta anamnesis que recoja los antecedentes de CCR o adenomas en el propio individuo o en familiares de primer grado (padres, hermanos e hijos), de segundo grado (abuelos, tíos y sobrinos) y de tercer grado (bisabuelos y primos)7,8.
En ausencia de antecedentes personales y/o familiares, la edad del individuo es la condición más influyente para determinar el riesgo de CCR.
Los individuos menores de 50 años, sin factores de riesgo adicionales, presentan un riesgo bajo para CCR, y no se consideran candidatos a cribado para esta patología (grado de recomendación A).
Los pacientes considerados de riesgo medio tienen más de 50 años, están asintomáticos y no presentan antecedentes personales de enfermedad inflamatoria intestinal ni antecedentes personales o familiares de CCR o pólipos adenomatosos. En estos pacientes, debe recomendarse el cribado anual o bienal mediante la detección de sangre oculta en heces y/o sigmoidoscopia cada 5 años, o colonoscopia cada 10 años (grado de recomendación A) (figura 1).
Se consideran de riesgo elevado aquellos individuos con factores de riesgo personal y/o familiar para el desarrollo de CCR (antecedentes personales de enfermedad inflamatoria intestinal, pólipos adenomatosos o antecedentes familiares de primer grado de CCR, poliposis adenomatosa familiar o de cáncer de colon hereditario sin poliposis [CCHSP]); son subsidiarios de programas de cribado o vigilancia específicos. Cuando en un individuo coexistan ambos tipos de factores (personales y familiares) la medida de prevención debe ir dirigida a la situación de mayor riesgo. En estos pacientes, la evidencia científica recomienda una búsqueda activa, ofrecerles el cribado con endoscopia, y evaluar la conveniencia de realizar análisis genéticos en unidades especializadas (grado de recomendación A). En la actualidad, una de las estrategias que más puede contribuir a disminuir las tasas de incidencia y mortalidad del CCR es la implantación de protocolos de seguimiento de los pólipos adenomatosos de riesgo (adenomas múltiples: ≥ 3, o adenomas avanzados: tubulares de tamaño ≥ 10 mm, vellosos o displasia de alto grado). Los estudios prospectivos han demostrado que la resección de los pólipos esporádicos y el posterior seguimiento colonoscópico de los adenomas de riesgo contribuye a reducir la incidencia de CCR en más de un 75%.
Cuando un individuo presenta clínica sospechosa del desarrollo de CCR no se considera tributario de cribado. En esta situación, debe realizarse una estrategia diagnóstica adecuada con el fin de confirmar o descartar esta patología. La extensión de la enfermedad en el momento del diagnóstico es el principal factor pronóstico8.
PRUEBAS DE CRIBADO
La eficacia del cribado en la población de riesgo medio está avalada científicamente (grado de recomendación A)5. No obstante, existe controversia sobre la estrategia de cribado más efectiva y con mejor relación coste-efectividad.
-
Las pruebas de detección de sangre oculta en heces (SOH) se llevan a cabo mediante el examen de dos muestras de cada una de las deposiciones de 3 días consecutivos. Se fundamentan en la emisión de sangre a la luz intestinal de algunos adenomas y del CCR, debido a la posible ulceración y friabilidad de éstos. Las pruebas de SOH con mejor evaluación y nivel de evidencia sobre su eficacia en la detección de neoplasias colorrectales con las que utilizan como reactivo el guayaco (grado de recomendación A). La principal limitación de su aplicación como prueba de cribado es el alto porcentaje de resultados falsos positivos (de un 2 a un 10%) que obliga a la realización posterior de una prueba más invasiva, como es la colonoscopia, con lo que un porcentaje de pacientes serán sometidos a una colonoscopia innecesaria. La especificidad de esta prueba para la detección de cualquier neoplasia colorrectal y para el CCR se aproxima al 80%. La sensibilidad es inferior para los adenomas ya que sangran con menor frecuencia. Otro inconveniente de esta prueba es que las restricciones dietéticas que precisa la detección de SOH basada en guayaco no reducen la tasa de resultados positivos y cuanto más se restringe la dieta, menor es el cumplimiento del cribado. Con el objetivo de mejorar la validez de la prueba y el cumplimiento de las restricciones dietéticas, se han desarrollado métodos inmunológicos cuantitativos y cuantitativos que detectan específicamente la hemoglobina humana, por lo que no precisan restricciones dietéticas durante la recogida de la muestra. Estos métodos inmunológicos mejoran la especificidad de la detección de CCR, pero pueden dar lugar a resultados falsos negativos en los sangrados procedentes del tracto gastrointestinal superior por el proceso de degradación de la hemoglobina en su recorrido por el tubo digestivo.
Otra alternativa podría consistir en la combinación de un método inmunológico tras la detección de un guayaco positivo, lo cual disminuye los resultados falsos positivos. La positividad de ambos métodos alcanza una sensibilidad para el CCR del 95,9% (IC 95%, 84,8-99,3%), y para las neoplasias colorrectales avanzadas del 87,8% (IC 95%, 80,1-92,9%). La Organización Mundial de la Salud (OMS) y la Organización Mundial de Endoscopia Digestiva recomiendan seleccionar los métodos basados en el guayaco cuando la adherencia de la población a la restricción dietética sea buena y se disponga de suficientes recursos endoscópicos para el seguimiento de pacientes con resultados positivos. En caso contrario, los métodos inmunológicos evitan los inconvenientes creados por las restricciones dietéticas y los fármacos, y suponen una mejora en la calidad del proceso. En España, las comunidades autónomas de Murcia, Cataluña, Valencia y Canarias disponen de programas piloto de cribado del CCR. Canarias y Murcia utilizan una prueba de cribado inmunológico mientras que las otras han empleado métodos basados en el guayaco, aunque planifican la migración hacia una prueba inmunológica9,10. - La sigmoidoscopia y la colonoscopia son pruebas más sensibles, pero también más invasivas y costosas. La sigmoidoscopia permite visualizar hasta 60 cm, con lo que se alcanza el recto, el sigma y la porción distal del colon descendente, donde se localizan el 60% de los CCR y la mayoría de los pólipos. La colonoscopia permite visualizar todo el colon, sobre todo cuando se efectúa bajo sedación. Ambas técnicas permiten realizar polipectomías. La ventaja atribuida a la colonoscopia se basa en que una proporción importante de las neoplasias avanzadas proximales presentan lesiones sincrónicas distales y, por tanto, no serían detectables mediante sigmoidoscopia.
- La combinación de SOH con guayaco y sigmoidoscopia no supera la eficacia de cada una de estas pruebas por separado, con cifras similares en la detección de neoplasias colorrectales avanzadas.
- Respecto al enema opaco, actualmente no hay datos científicos que avalen su eficacia como prueba en el cribado poblacional. No obstante, podría ser el procedimiento indicado en aquellos pacientes en los que la colonoscopia resultase especialmente peligrosa debido a la existencia de otras patologías médicas concomitantes (trastornos cardiopulmonares o situaciones de anticoagulación). El inconveniente principal es que el paciente tiene que someterse a una colonoscopia posterior si se identifican anomalías durante la exploración.
- La limitación más importante en la realización de un cribado poblacional se encuentra en la participación de la población, especialmente cuando se compara con otros programas. Esta baja participación puede explicarse por diferentes motivos: las características de las distintas pruebas de cribado, así como la preparación previa que muchas de ellas requieren, el insuficiente conocimiento tanto del cribado como de la enfermedad que la población tiene, y la baja percepción social de sus beneficios.
- Recientemente se están evaluando otras pruebas de cribado poblacional como la colonoscopia virtual o colonografía por tomografía computarizada (TC). Se trata de un examen radiológico que emplea la TC para obtener una visión interna del colon. Sus principales indicaciones son la detección de pólipos en el colon; el estudio de pacientes cuyas condiciones clínicas pueden suponer un aumento del riesgo de complicaciones en la colonoscopia convencional, como tratamientos anticoagulantes o dificultades respiratorias; y otra indicación sería completar el estudio en colonoscopias incompletas cuando no se puede completar el estudio mediante la realización de una colonoscopia convencional, porque el intestino se ha estrechado o está obstruido por cualquier causa, como por ejemplo un tumor, y también cuando la colonoscopia convencional no puede alcanzar la longitud total del colon, lo que ocurre hasta en el 10% de las ocasiones. Carece de los riesgos de la colonoscopia óptica, y los pacientes no requieren sedación, pero se ha desaconsejado por tratarse sólo de un procedimiento diagnóstico ya que no permite la resección simultánea de los pólipos, ni la visualización de los de tamaño inferior a 6 mm. Además, si el resultado es patológico, habría que realizar una colonoscopia convencional, idealmente el mismo día o al siguiente, para evitar una nueva preparación intestinal13.
Otra nueva prueba que se está evaluando es la detección de mutaciones del ADN en heces, que permite detectar carcinomas no sangrantes, mejorando así teóricamente la sensibilidad de las pruebas de SOH.
- Se recomienda el cribado de CCR en las personas de riesgo medio a partir de los 50 años, con alguna de las siguientes pruebas: SOH con periodicidad anual o bienal y/o sigmoidoscopia cada 5 años o colonoscopia cada 10 años. Los programas poblacionales deberían cubrir a la población de 50 a 74 años utilizando como método de cribado una prueba de SOH con técnicas inmunológicas cuantitativas, aplicada con intervalo bienal. Si el cribado es oportunista, la prueba de primera elección es también la de SOH inmunológica, aunque también son válidas la de SOH con guayaco, la sigmoidoscopia o la colonoscopia, dependiendo de su disponibilidad y aceptabilidad10,11.
CRIBADO EN INDIVIDUOS DE RIESGO ELEVADO
Cáncer colorrectal familiar
El CCR familiar, llamado así para diferenciarlo de las formas inequívocamente hereditarias, representa un 25-30% del total de los casos de cáncer de colon y recto. Se desconoce exactamente el mecanismo responsable de esta agregación familiar, aunque probablemente se trate de una alteración genética multifactorial. El número de familiares afectados, el grado de parentesco y la edad de diagnóstico del CCR en el familiar son las variables asociadas a un mayor riesgo de presentar CCR en los distintos estudios realizados. El riesgo de presentar esta neoplasia en individuos con familiares de primer grado que han padecido CCR es 2-3 veces superior al de la población general. Varios estudios demuestran que este riesgo es mayor cuando el familiar afectado es un hermano que cuando es un progenitor. El riesgo de CCR en individuos con familiares de primer grado afectados disminuye con la edad; a los 70 años este riesgo todavía es superior al de la población sin familiares afectados. La existencia de familiares de segundo grado (abuelos, tíos y sobrinos) o tercer grado (bisabuelos y primos) afectados de CCR también se ha asociado a un incremento del riesgo de presentar esta neoplasia. El antecedente familiar de cáncer de colon comporta un mayor riesgo que el de cáncer de recto. Asimismo, los individuos con antecedentes familiares de adenoma colorrectal tienen un mayor riesgo de desarrollar CCR. En aquéllos con antecedentes familiares de CCR se debería iniciar el cribado mediante colonoscopia a partir de los 40 años de edad, o 10 años antes de la edad de diagnóstico del familiar afectado más joven (figura 2)5,10.
Poliposis adenomatosa familiar
La poliposis adenomatosa familiar (PAF) es una enfermedad hereditaria autosómica dominante causada por una mutación del gen APC, que se caracteriza por la presencia de múltiples pólipos adenomatosos colónicos con elevado riesgo de transformación neoplásica. Su incidencia se estima en 1 por 10.000 habitantes, afecta a ambos sexos por igual y representa menos del 1% de la totalidad de casos de CCR. Los pacientes con PAF y sus familiares deben ser remitidos a unidades especializadas en CCR hereditario para su registro y atención. El análisis del gen APC debe considerarse para confirmar el diagnóstico de PAF y realizar el diagnóstico presintomático de los familiares con riesgo. En los individuos con riesgo de PAF clásica (portadores de mutaciones en el genAPC y pertenecientes a familias que cumplen los criterios clínicos en las que se ha identificado la mutación causal) se debe realizar una sigmoidoscopia cada 1-2 años a partir de los 13-15 años y hasta los 40 años de edad, y cada 5 años hasta los 50-60 años de edad. En los individuos con riesgo de PAF atenuada debe realizarse una colonoscopia cada 1-2 años a partir de los 15-25 años, en función de la edad de presentación de la enfermedad en los familiares afectados. Una vez detectada la presencia de adenomas, debe realizarse una colonoscopia anual hasta la realización del tratamiento definitivo. Un 40% de los pacientes con PAF presenta manifestaciones extracolónicas asociadas, y las más frecuentes son las lesiones gastroduodenales (hipertrofia glandular fúndica, adenomas o pólipos hiperplásicos, y adenocarcinoma). El cribado de las manifestaciones gastroduodenales contempla la realización de una endoscopia gastroduodenal cada 4-5 años a partir de los 25-30 años de edad5,10.
Cáncer colorrectal hereditario no asociado a poliposis (CCHNP) o síndrome de Lynch
El CCHNP es una enfermedad hereditaria con patrón autosómico dominante debida a mutaciones germinales en genes reparadores de errores de replicación del ADN. A pesar de tratarse de la forma de CCR hereditario más frecuente, en la actualidad se acepta que esta entidad representa entre el 0,9 y el 2% del total de casos de CCR. El diagnóstico clínico de CCHNP se establece a partir de la historia familiar, y su definición se basa en los criterios de Amsterdam II (tabla 1), en los que tienen un mayor protagonismo las neoplasias extracolónicas. Clínicamente, se caracteriza por el desarrollo temprano de CCR, habitualmente antes de los 50 años de edad, de predominio en el colon derecho y con una elevada tendencia a presentar neoplasias sincrónicas, ya sea en el propio colon y el recto, o en otros órganos (endometrio, estómago, sistema urinario, ovarios, vías biliares, intestino delgado). La neoplasia extracolónica más frecuente es la de endometrio. El análisis de los genes reparadores del ADN permite confirmar el diagnóstico de síndrome de Lynch y realizar el diagnóstico presintomático en los familiares de riesgo. El cribado endoscópico en los individuos con riesgo pertenecientes a familias con síndrome de Lynch reduce la incidencia y la mortalidad por CCR; este cribado es coste-efectivo. En los individuos con riesgo de padecer síndrome de Lynch debe realizarse una colonoscopia cada 1-2 años a partir de los 20-25 años de edad, o 10 años antes de la edad de diagnóstico del familiar afectado más joven (lo primero que ocurra). El cribado de las neoplasias extracolónicas en el síndrome de Lynch debería individualizarse en función de la predisposición familiar para una determinada neoplasia. En las mujeres con riesgo pertenecientes a familias con síndrome de Lynch se debería valorar el cribado de cáncer de endometrio mediante ultrasonografía transvaginal y/o aspirado/biopsia endometrial con periodicidad anual a partir de los 30-35 años de edad. En individuos con riesgo pertenecientes a familias con síndrome de Lynch y cáncer gástrico asociado debería valorarse la realización de una gastroscopia cada 1-2 años a partir de los 30-35 años de edad. En los familiares de los pacientes con este síndrome y neoplasias urinarias asociadas debería realizarse una ultrasonografía y citología urinaria cada 1-2 años a partir de los 30-35 años de edad. En los pacientes pertenecientes a familias con síndrome de Lynch que desarrollan CCR se debería valorar la realización de una resección extensa (colectomía o proctocolectomía total) como prevención del desarrollo de otras neoplasias posteriormente5,10.
Adenomas colorrectales
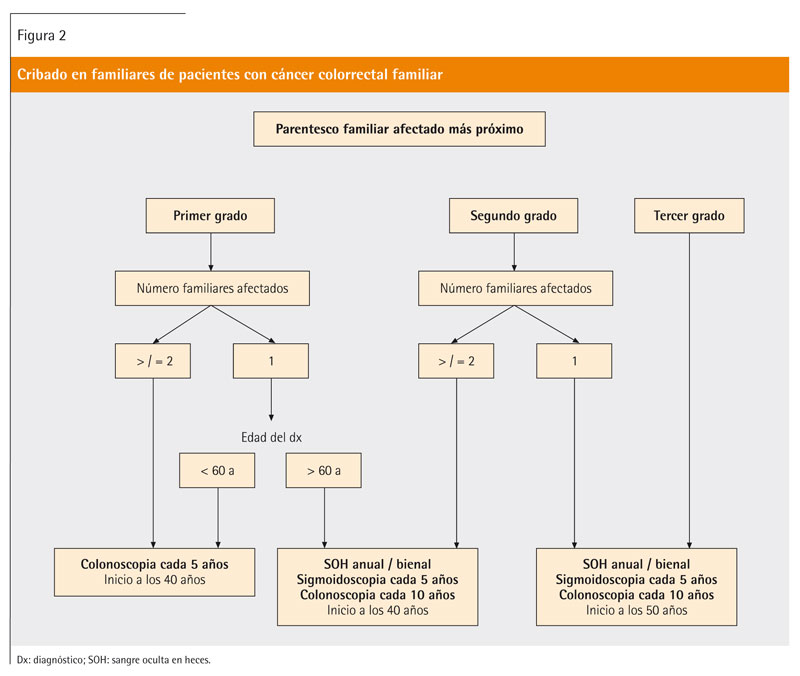
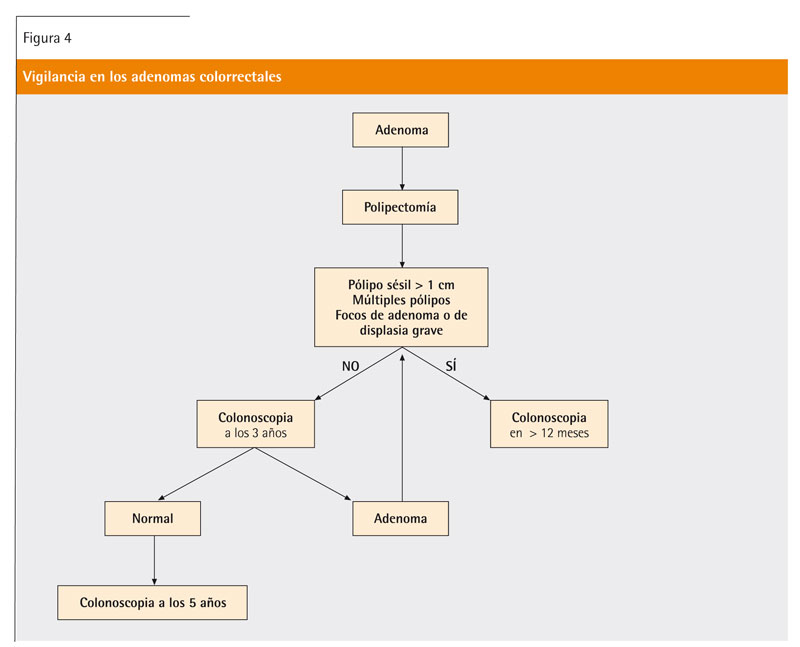
La mayoría de los CCR se inician a partir de un adenoma y, aunque no todos evolucionen a cáncer, esta lesión debería ser considerada como premaligna (ver figura 3). Los pólipos neoplásicos incluyen los adenomas, y los no neoplásicos los pólipos hiperplásicos, inflamatorios y hamartomatosos. La mayoría de los pólipos colorrectales son esporádicos y corresponden a adenomas (67%) y a pólipos hiperplásicos (11%). El adenoma con displasia de alto grado (antiguamente denominado carcinoma in situ) se considera una neoplasia sin capacidad de diseminación. Cuando las áreas de transformación carcinomatosa se extienden a la capa submucosa se considera un carcinoma invasivo con capacidad de diseminación. La presencia de displasia de alto grado se asocia con el tamaño de la lesión, la proporción de componente velloso y la edad del paciente. En los pacientes con adenoma colorrectal con displasia de bajo o alto grado, o carcinoma no invasivo, la polipectomía endoscópica se considera el tratamiento definitivo. En aquellos con adenoma colorrectal con áreas de carcinoma que invaden la submucosa se considera que la polipectomía endoscópica es el tratamiento definitivo cuando la resección es completa y en bloque, y se cumplen todos los criterios de buen pronóstico (margen de resección libre de enfermedad, carcinoma bien o moderadamente diferenciado y ausencia de invasión vascular y linfática). En los adenomas sésiles de gran tamaño y con base de implantación amplia el tratamiento inicial debe ser la resección quirúrgica. La vigilancia pospolipectomía permite la detección tanto de lesiones residuales o sincrónicas que pasaron desapercibidas en la exploración basal como de lesiones metacrónicas (figura 4). Las estrategias de cribado deben ir dirigidas a detectar de forma temprana los adenomas avanzados (lesiones ≥ 10 mm, con componente velloso o con displasia de alto grado). Todos los pólipos identificados durante la endoscopia deben ser resecados, ya sea mediante polipectomía endoscópica o quirúrgica. Si la colonoscopia únicamente demuestra la presencia de pólipos hiperplásicos rectales de pequeño tamaño, esta exploración se considera normal y, por tanto, estos pacientes deben ser incluidos en el cribado poblacional transcurridos 10 años. Los pacientes con adenoma con áreas de carcinoma que invaden la submucosa a los que se les ha realizado una polipectomía endoscópica deberían ser reexaminados en un período de 3 meses mediante colonoscopia y toma de biopsias con el fin de confirmar la resección completa de la lesión. Los pacientes con un adenoma sésil grande en los que se realiza una resección endoscópica fragmentada deben ser reexaminados en un plazo de 3-6 meses mediante colonoscopia y toma de biopsias con el fin de confirmar la resección completa de la lesión. En aquellos otros con más de 10 adenomas en una exploración debería realizarse una colonoscopia en un intervalo inferior a 3 años, y descartar la presencia de un síndrome de poliposis familiar. En los pacientes con 3-10 adenomas o un adenoma avanzado (≥ 10 mm, con componente velloso, o con displasia de alto grado), la primera colonoscopia de vigilancia debe efectuarse a los 3 años de la exploración basal, mientras que en los que tienen 1 o 2 adenomas tubulares pequeños (< 10 mm), ésta puede demorarse hasta los 5 o 10 años5,10.
Enfermedad inflamatoria intestinal
Los pacientes con colitis ulcerosa y enfermedad de Crohn con afectación colónica presentan un riesgo aumentado de desarrollar CCR. En la enfermedad inflamatoria intestinal (EII) el riesgo aumenta con la duración y la extensión de la enfermedad, la coexistencia de colangitis esclerosante primaria, la presencia de antecedentes familiares de esta neoplasia y de seudopólipos postinflamación. No hay estudios que proporcionen pruebas científicas inequívocas a favor de la vigilancia endoscópica en pacientes con EII. El diagnóstico temprano de la displasia es, por el momento, el mejor marcador de riesgo de CCR en estos pacientes. El tratamiento de las lesiones precancerosas mediante polipectomía endoscópica y, en ocasiones, cirugía, es eficaz para la prevención del CCR12,14.
LECTURAS RECOMENDADAS
Guía de referencia rápida de prevención del cáncer colorrectal. Asociación Española de Gastroenterología. Sociedad Española de Medicina de Familia y Comunitaria (semFYC) (Actualización 2009). La guía completa está disponible en:http://www.guiasgastro.net
Esta guía proporciona un resumen de las principales recomendaciones de laGuía de práctica clínica sobre el manejo del paciente con cáncer colorrectal.
Instituto Nacional del Cáncer de Estados Unidos. Disponible en: http://www. cancer.gov/espanol/pdq/tratamiento/colon/HealthProfessional
Página web del Instituto Nacional del Cáncer de Estados Unidos para la investigación y la documentación oncológica, con una página dedicada al cáncer colorrectal. Presenta versiones de acceso libre dirigidas a los pacientes y profesionales de la salud, tanto en español (con menos contenido), como en inglés.
BIBLIOGRAFÍA
- Abraldes Belchiarelli A, Santamaría Rodríguez G, Rodríguez Ramos C. Tumores de intestino grueso. Medicine.2008;10(7):442-8.
- Ferlay J, Bray F, Pisani P, Parkin DM. GLOBOCAN 2002: Cancer incidence, mortality and prevalence worldwide. IARC Cancer Database Nº 5, version 2.0. Lyon: IARCPress; 2006. Disponible en: http://www.dep.iarc.Fr
- Key TJ, Schatzkin A, Wilett WC, et al. Diet, nutrition and the prevention of cancer. Public Health Nutr. 2006;7:187-200.
- Nieto Pol E, Álvarez García AJ, Muñoz García JL. Colonoscopia en la prevención y el cribado del cáncer colorrectal. FMC. 2004;11(7):383-93.
- Guía de referencia de Prevención del Cáncer colorrectal (Actualización 2009). Disponible en: http:www.guiasgastro.net
- Asociación Española de Gastroenterología (AEG). Disponible en: http://www.aegastro.es
- Scholefield JH. ABC of colorectal cancer. Screening. BMJ. 2004;321:1004-6.
- Soetikno RM, Kaltenbach T, Rouse RV, et al. Prevalence of nonpolypoid (flat and depressed) colorectal neoplasms in asymptomatic and symptomatic adults. J Am Med Assoc. 2008;299(9):1027-35.
- Walsh JM, Terdiman JP. Colorectal cancer screening: clinical applications. JAMA. 2003;289:1297-302.
- Hewitson P, Glasziou P, Irwig L, et al. Detección del cáncer colorrectal con la prueba de sangre oculta en materia fecal (Hemoccult) (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus. 2008. Número 2. Oxford: Update Software Ltd. Disponible en: http://www.update-software.com (Traducida de The Cochrane Library.2008 Issue 2. Chichester. UK: John Wiley & Sons Ltd.)
- Burch JA, Soares-Weiser K, St John DJ, et al. Diagnostic accuracy of faecal occult blood tests used in screening for colorectal cancer: A systematic review. J Med Screen. 2007;14(3):132-7.
- Marzo Castillejo M, Piñol V, Mascort JJ, et al. Estrategias de cribado en el cáncer colorrectal. FMC. 2005;12(8):527-35.
- Pickhardt PJ, Choi JR, Hwang I, et al. Computed tomographic virtual colonoscopy to screen for colorectal neoplasia in asymptomatic adults. N Engl J Med. 2003;349(23):2191-200.
- Marzo Castillejo M, Bellas Beceiro B, Melus Palazón E, et al. Prevención del cáncer. Programa de actividades preventivas y de promoción de la salud (Actualización 2009). semFYC. 2009. 126-30.
Luis Emilio 25-02-16
Excelente