




Resumen
En cuanto a nuevos fármacos, hemos encontrado tres: el lesinurad, indicado como tratamiento adyuvante de la hiperuricemia, que no ha supuesto ningún avance terapéutico; la propiverina, como tratamiento sintomático de la incontinencia urinaria o del incremento de la frecuencia y urgencia urinarias en pacientes adultos con síntomas de vejiga hiperactiva, que tampoco ha supuesto un avance terapéutico y, por último, la prasterona, que está indicado en mujeres posmenopáusicas con síntomas moderados o graves de atrofia vulvovaginal; los estudios no han podido determinar si es mejor opción que los estrógenos vaginales en dosis bajas .
En cuanto a las últimas alertas recogidas, nos encontramos con el conocido efecto del metamizol y la agranulocitosis, las nuevas restricciones de las quinolonas, el riesgo de hidroclorotiazida que podría aumentar el riesgo de cáncer cutáneo y que, en Francia, los medicamentos para la enfermedad de Alzheimer dejan de ser reembolsables.
Nuevos medicamentos a exámen
LESINURAD (Zurampic®)
Indicaciones: en combinación con un inhibidor de la xantina oxidasa, para el tratamiento adyuvante de la hiperuricemia en pacientes adultos con gota (con o sin tofos) que no han alcanzado las concentraciones séricas de ácido úrico deseadas con una dosis adecuada de un inhibidor de la xantina oxidasa en monoterapia.
Posología: 200 mg/día, administrados por la mañana junto con la dosis del inhibidor de la xantina oxidasa y alimentos. La ficha técnica recomienda asegurar una ingestión hídrica adecuada (2 L/día) para mitigar el riesgo de acontecimientos renales. Lesinurad no debe usarse en monoterapia y debe suspenderse si se interrumpe el tratamiento de la xantina oxidasa.
No es necesario realizar ajustes de dosis en pacientes de edad avanzada o con insuficiencia renal o hepática leve o moderada.
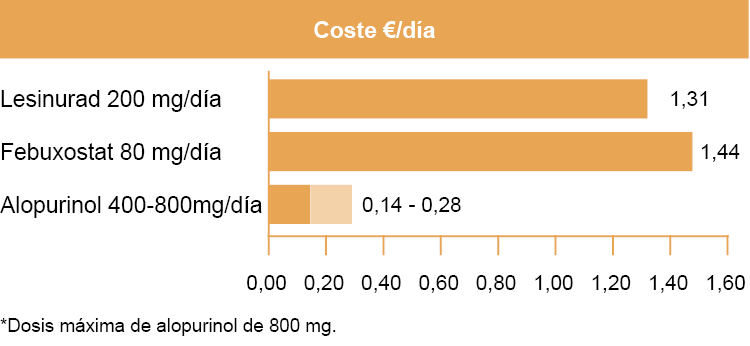
Coste:
Eficacia: se ha evaluado en tres ensayos clínicos pivotales fase II, aleatorizados, doble ciego y controlados con placebo, en comparación con alopurinol (CLEAR1 y CLEAR2) o febuxostat (CRYSTAL).
En combinación con alopurinol: obtuvo resultados estadísticamente significativos en comparación con alopurinol en monoterapia, para la variable principal (niveles plasmáticos de ácido úrico <6 mg/dL). Aunque no se obtuvieron diferencias significativas para las variables secundarias, es decir tasa de crisis agudas de gota que requirieron tratamiento (meses 6 y 12) y proporción de pacientes que experimentaron resolución completa de al menos un tofo (mes 12).
En combinación con febuxostat: no demostró ser superior a febuxostat en monoterapia con respecto a su variable principal (proporción de pacientes con niveles plasmáticos de ácido úrico <5 mg/dL a los 6 meses). Para el objetivo secundario (resolución de tofos) demostró ser más eficaz que febuxostat en monoterapia.
Seguridad: entre los efectos secundariosdestacan la infección tipo influenza, el reflujo gastroesofágico, la cefalea y el aumento de creatinina plasmática.
Precauciones: los datos disponibles no permiten descartar un impacto negativo en la seguridad cardiovascular ni se ha podido establecer una relación causal con lesinurad. El laboratorio titular está obligado a realizar un estudio postautorización que evalúe la seguridad cardiovascular, que está previsto que finalice en 2019. No se recomienda usar lesinurad en pacientes con insuficiencia renal grave (aclaramiento de creatinina [CrCl] <30 mL/min) o terminal, trasplantados renales o hemodializados y se debe utilizar con precaución en la insuficiencia renal moderada (CrCl 30-45 mL/min). No se dispone de datos de morbimortalidad a largo plazo. Está contraindicado en los síndromes de lisis tumoral y de Lesch-Nyhan.
Interacciones: sustratos de CYP3A, sustratos del CYP2B6, salicilatos, diuréticos tiazídicos, warfarina, anticonceptivos hormonales, inhibidores e inductores de CYP2C9, rifampicina, inhibidores de la epóxido hidrolasa.
Lugar en la terapéutica: está indicado en combinación de un inhibidor de la xantina-oxidasa para el tratamiento de la hiperuricemia en pacientes adultos con una afección sintomática relevante y cuya hiperuricemia no ha respondido adecuadamente con alopurinol o febuxostat en dosis máximas toleradas, restringiéndose su uso combinado con febuxostat únicamente cuando se haya utilizado previamente en combinación con alopurinol o cuando este esté contraindicado.
Para ampliar información:
https://ec.europa.eu/health/documents/community-register/2016/20160218134007/anx_134007_es.pdf
PROPIVERINA (Mictonorm®)
Indicaciones: tratamiento sintomático de la incontinencia urinaria o incremento de la frecuencia y urgencia urinarias en pacientes adultos con síndrome de vejiga hiperactiva.
Posología: 30 mg una vez al día con o sin alimentos. No se requiere ajuste de dosis en personas de edad avanzada. Se recomienda reevaluar la eficacia del medicamento a partir de las 4 semanas.
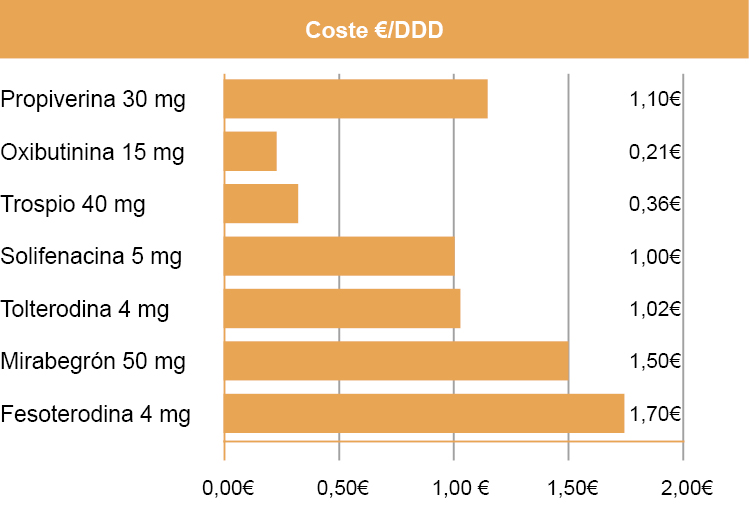
Coste:
Coste comparativo. Fuente: Osakidetza/Cevime, marzo de 2018.
Eficacia: se estudió la eficacia de la propiverina de liberación modificada frente a propiverina de liberación inmediata (no comercializada en España) y placebo. Se realizó un ensayo multicéntrico, aleatorizado, doble ciego, doble enmascaramiento, con grupos paralelos durante 32 días. En este estudio, la propiverina de liberación modificada mostró reducir, comparando con placebo, los episodios de incontinencia en 24 horas con una diferencia en las medias de 0,77 (IC 95 %: 0,44 a 1,10) y disminuciones estadísticamente significativas en las variables secundarias, número de episodios de urgencia en 24 horas y número de micciones en 24 horas.
Asimismo, también se estudió la eficacia de propiverina de liberación modificada frente a tolterodina en un ensayo de no inferioridad, multicéntrico, aleatorizado, doble ciego, doble enmascaramiento, en grupos paralelos, de 8 semanas. Demostró no ser inferior en el número de micciones en 24 horas, con una diferencia en las medias de -0,55 (IC 95 %: -1,3 a 0,2). En variables secundarias, se observó una mayor disminución en el número de episodios de incontinencia de urgencia con propiverina.
Seguridad:entre los efectos secundarioscabe destacar la sequedad de boca (22-28 %), la cefalea, los trastornos de la acomodación, la discapacidad visual, el estreñimiento, el dolor abdominal, la dispepsia y la fatiga.
Precauciones: no se recomienda en pacientes con insuficiencia hepática severa-moderada. Precaución en pacientes con neuropatía autonómica. Pueden agravarse los síntomas de la insuficiencia cardíaca congestiva severa (NYHA IV), hipertrofia prostática, hernia de hiato con reflujo gastroesofágico, arritmia cardíaca, taquicardia, y glaucoma agudo de ángulo cerrado.
Antes del tratamiento, se debe descartar que los síntomas sean debidos a una lesión renal o a insuficiencia cardíaca congestiva o a patologías orgánicas de la vejiga.
Interacciones: antidepresivos tricíclicos, benzodiazepinas, anticolinérgicos, amantadina, neurolépticos beta-simpaticomiméticos, fármacos colinérgicos, isoniazida, metoclopramida, fármacos que se metabolizan a través del CYP3A4, y metamizol. No hay estudios farmacocinéticos en pacientes que reciben tratamiento concomitante con inhibidores potentes del CYP3A4.
Lugar en la terapéutica: el objetivo del tratamiento de la vejiga hiperactiva es mejorar la calidad de vida de los pacientes al disminuir los síntomas de urgencia, la frecuencia y la incontinencia urinaria. El tratamiento inicial para todos los pacientes incluye cambios en el estilo de vida y tratamiento conductual. Los anticolinérgicos urinarios son una alternativa más en el tratamiento, cuando las medidas no farmacológicas no han sido suficientes, y en combinación con estas. Sus beneficios respecto a placebo son pequeños y la eficacia entre ellos es similar. Se debe destacar la baja adherencia observada en estos fármacos al año de tratamiento (17-35 %), motivada por su modesta eficacia y sus efectos adversos.
Para ampliar información:
http://www.navarra.es/NR/rdonlyres/6DFC5382-F45B-4757-99F3-899CBF1B9AD5/425572/FET_2018_1.pdf
PRASTERONA (Intrarosa®)
Indicaciones: es un esteroide indicado para el tratamiento de la dispareunia moderada a severa en el contexto de la atrofia vulvar o vaginal secundaria a la menopausia. La prasterona también es conocida como deshidorepiandrosterona (DHEA).
Posología: óvulo vaginal de 6,5 mg una vez al día.
Coste: autorizado, pendiente de comercializar.
Eficacia: en un ensayo aleatorizado de mujeres posmenopáusicas (n = 482) con dispareunia, se asignó prasterona 6,5 mg vaginales dosis día o placebo durante 12 semanas. Demostró mejores puntuaciones generales en el Índice de Función Sexual Femenina y en los dominios de dolor y satisfacción en el grupo prasterona (DHEA). A las 12 semanas, el tratamiento con DHEA vaginal produjo un aumento significativo desde el inicio en el suero de DHEA, testosterona y estrona, pero no en los niveles de estradiol, aunque, como se señaló en el resumen del estudio, la DHEA, el estradiol, la estrona y la testosterona permanecieron dentro del rango habitual de una mujer posmenopáusica.
En otros dos estudios en los que participaron 813 mujeres posmenopáusicas con atrofia vulvar, vaginal, o ambas, se observó que el tratamiento con prasterona 6,5 mg día, era más eficaz que placebo para reducir los signos de adelgazamiento de los tejidos vaginales.
Seguridad: Entre los efectos secundarios, el más frecuente (10 %) es la secreción vaginal.
Precauciones: las pequeñas elevaciones de estrona en mujeres tratadas con prasterona aumentan la preocupación por el uso de este tratamiento en mujeres con o en riesgo de neoplasias malignas sensibles al estrógeno, en particular aquellas con cáncer de mama que reciben tratamiento con un inhibidor de la aromatasa.
No debe utilizarse en las siguientes condiciones: hemorragia genital en la que no se ha diagnosticado la causa, presencia o sospecha de cáncer de mama o de cáncer dependiente de estrógenos, cáncer de mama previo, hiperplasia endometrial no tratada, enfermedad hepática aguda, antecedentes o presencia de tromboembolia venosa, trastornos trombofílicos, enfermedad tromboembólica arterial activa o reciente, porfiria.
Lugar en la terapéutica: para la mayoría de las mujeres con atrofia vulvovaginal sintomática que no mejoran con lubricantes y humectantes vaginales y que son candidatas a terapia hormonal, sugerimos una terapia con estrógenos vaginales en dosis bajas en lugar de la DHEA vaginal. Hay más experiencia clínica y datos con respecto al estrógeno vaginal y es probable que la dosis dos veces por semana sea más fácil para la mayoría de las pacientes. La prasterona es efectiva, pero se asocia con un ligero aumento en los niveles de DHEA, testosterona y estrona circulantes, y su eficacia no se ha comparado directamente con el estrógeno vaginal. Por otro lado, la prasterona se ha estudiado como un medicamento potencial para aumentar la libido en mujeres posmenopáusicas, pero su eficacia es incierta y es poco probable que los niveles séricos de DHEA con esta dosis de prasterona vaginal tengan un impacto. La libido puede mejorar con el tratamiento adecuado de la dispareunia.
Para ampliar información
https://www.ema.europa.eu/documents/overview/intrarosa-epar-summary-public_es.pdf
https://www.aemps.gob.es/cima/publico/detalle.html
Alertas y seguridad farmacológicas
METAMIZOL Y RIESGO DE AGRANULOCITOSIS
Metamizol es un analgésico y antipirético comercializado desde hace más de 50 años en España con diferentes nombres comerciales. Se trata de medicamentos muy utilizados, indicados como analgésicos en distintas situaciones que cursan con dolor agudo moderado a intenso, y como antipirético cuando otras alternativas no son eficaces. Aunque se ha discutido desde hace años sobre una mayor susceptibilidad para la agranulocitosis en la población del norte de Europay se han estudiado ciertos factores genéticos, con la información disponible no se puede ni descartar ni confirmar un mayor riesgo en poblaciones con características étnicas específicas.
A partir de los datos de consumo de metamizol y los casos de agranulocitosis notificados, la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) recuerda que estos medicamentos están catalogados como de prescripción y recomienda a los profesionales sanitarios:
- Utilizar metamizol solo para tratamientos de corta duración (7 días como máximo), dentro de sus indicaciones autorizadas y en las dosis mínimas eficaces.
- Si es necesario un tratamiento más prolongado, realizar controles hematológicos periódicos, incluyendo la fórmula leucocitaria.
- Durante el tratamiento, vigilar la aparición de sintomatología indicativa de agranulocitosis, informando a los pacientes para que, en tal caso, suspendan el tratamiento.
- Antes de prescribir metamizol, realizar una anamnesis detallada para evitar su uso en pacientes con antecedentes de reacciones de hipersensibilidad o hematológicas a metamizol, pacientes con tratamiento inmunosupresor o con medicamentos que pueden producir agranulocitosis.
- Adoptar especial precaución en caso de pacientes de edad avanzada.
- No utilizar metamizol en pacientes en los no sea posible realizar controles (por ejemplo, población flotante).
Para ampliar información
QUINOLONAS Y FLUOROQUINOLONAS DE ADMINISTRACIÓN SISTÉMICA: NUEVAS RESTRICCIONES DE USO
Las quinolonas y fluoroquinolonas (ciprofloxacino, levofloxacino, moxifloxacino, norfloxacino, ofloxacino y ácido pipemídico) son antibióticos sintéticos utilizados para el tratamiento de un amplio espectro de infecciones bacterianas entre las que se incluyen infecciones de las vías urinarias y respiratorias, del aparato genital y gastrointestinal, así como infecciones cutáneas, óseas y articulares.
Recientemente, el Comité para la Evaluación de Riesgos en Farmacovigilancia (PRAC, del inglés Pharmacovigilance Risk Assessment Committee) europeo ha evaluado el impacto que las reacciones adversas incapacitantes, de duración prolongada, y potencialmente irreversibles que afectan a los sistemas nervioso y osteomuscular, podían ocasionar sobre la relación beneficio-riesgo de este grupo farmacológico.
Las principales conclusiones fueron las siguientes:
- Las reacciones adversas osteomusculares y del sistema nervioso, incapacitantes, de duración prolongada, y potencialmente irreversibles, aunque se consideran poco frecuentes, afectan a todas las quinolonas y fluoroquinolonas, constituyendo un efecto de clase.
- Debido a la gravedad de las reacciones observadas y teniendo en cuenta que pueden producirse en personas previamente sanas, cualquier prescripción de antibióticos quinolónicos o fluoroquinolónicos deberá realizarse tras una cuidadosa valoración de su relación beneficio-riesgo.
- Para los pacientes con infecciones graves por bacterias sensibles, este tipo de antibióticos continúa siendo una importante opción terapéutica.
- Por el contrario, en el caso de infecciones leves o autolimitadas, los beneficios de este tratamiento no superan el riesgo de sufrir las reacciones adversas mencionadas.
- Puesto que el ácido nalidíxico, cinoxacina, flumequina (no comercializados en España) y el ácido pipemídico, no conservan ninguna indicación para la que el balance beneficio-riesgo resulte favorable, se recomienda suspender su autorización de comercialización en toda la Unión Europea.
Según lo anterior, y hasta que esta decisión no sea efectiva, se recomienda a los profesionales sanitarios:
-
No prescribir antibióticos quinolónicos ni fluoroquinolónicos:
- Para el tratamiento de infecciones leves o autolimitadas.
- Para realizar profilaxis de la diarrea del viajero o de las infecciones recurrentes de vías urinarias bajas.
- A pacientes con antecedentes de reacciones adversas graves tras la administración de este tipo de antibióticos.
- Utilizar quinolonas o fluoroquinolonas para el tratamiento de infecciones leves o moderadamente graves exclusivamente cuando otros antibióticos recomendados no resulten eficaces o no sean tolerados.
- Tener en cuenta a la hora de prescribir, que los pacientes de edad avanzada, trasplantados o aquellos en tratamiento con corticoides, presentan un mayor riesgo de sufrir lesiones tendinosas.
- Indicar a los pacientes que interrumpan el tratamiento con este tipo de antibióticos y acudan al médico en caso de que presenten reacciones adversas de tipo osteomuscular o del sistema nervioso anteriormente mencionadas.
Por otro lado, en una comunicación enviada de los laboratorios a los profesionales sanitarios, se informa de que las fluoroquinolonas sistémicas e inhaladas pueden aumentar el riesgo de aneurisma y disección aórtica,especialmente en las personas de edad avanzada. Los estudios epidemiológicos describen un aumento en el riesgo de aneurisma y disección aórtica de, aproximadamente, el doble en los pacientes tratados con fluoroquinolonas sistémicas en comparación con los que no toman ningún antibiótico o toman otros tipos de antibiótico (amoxicilina).
Por ello, advierten:
- Las fluoroquinolonas sistémicas e inhaladas pueden aumentar el riesgo de aneurisma y disección aórtica, especialmente en personas de edad avanzada.
- En los pacientes con riesgo de aneurisma y disección aórtica, las fluoroquinolonas solo deben utilizarse después de una minuciosa evaluación riesgo/beneficio y tras considerar otras opciones terapéuticas previamente.
- Entre los factores que predisponen al aneurisma y disección aórtica se encuentran los antecedentes familiares de aneurisma, aneurisma o disección aórtica preexistente, síndrome de Marfan, síndrome de Ehlers-Danlos vascular, arteritis de Takayasu, arteritis de células gigantes, enfermedad de Behçet, hipertensión y aterosclerosis.
- Se debe advertir a los pacientes del riesgo de aneurisma y disección aórtica e informar que deben acudir a urgencias en caso de presentar dolor abdominal, de pecho o espalda, de naturaleza súbita/aguda.
Para ampliar información
https://sinaem.agemed.es/CartasFarmacovigilanciaDoc/2018/2018-10-23-Fluoroquinolonas-DHPC_ES.pdf
HIDROCLOROTIAZIDA: EL USO CONTINUO Y PROLONGADO EN EL TIEMPO PODRÍA AUMENTAR EL RIESGO DE CÁNCER CUTÁNEO NO MELANOCÍTICO
Los resultados de dos estudios epidemiológicos realizados en Dinamarca indican un posible aumento del riesgo de desarrollar carcinoma basocelular y espinocelular en pacientes sometidos a tratamientos continuados y prolongados con hidroclorotiazida, que podría explicarse por su acción fototóxica.
Tras la revisión realizada de los dos estudios mencionados, así como del resto de información disponible procedente de la bibliografía médica, elComité para la Evaluación de Riesgos en Farmacovigilancia europeoha concluido lo siguiente:
- Según los estudios daneses, en pacientes expuestos a hidroclorotiazida, con dosis acumuladas de 50 000 mg o superiores, el riesgo de carcinoma basocelular podría incrementarse en 1,3 veces y el riesgo de carcinoma espinocelular, en 4 veces. Dosis acumuladas superiores se asociaban con un riesgo mayor. Una dosis acumulada de 50 000 mg correspondería, por ejemplo, al uso diario de 12,5 mg de hidroclorotiazida durante, aproximadamente, 11 años.
- Existe un mecanismo biológico plausible que podría explicar este aumento de riesgo ya que este principio activo tiene actividad fotosensibilizante.
- No se ha observado un incremento en el riesgo de desarrollar cáncer de piel de tipo melanocítico.
La AEMPS está llevando a cabo un estudio con datos procedentes de la base de datos BIFAP con objeto de obtener información sobre esta asociación en la población española. Mientras tanto, la AEMPS recomienda a los profesionales sanitarios:
- Reconsiderar el uso de hidroclorotiazida en pacientes con antecedentes de cáncer de piel no melanocítico.
- Solicitar una valoración especializada en caso de lesiones cutáneas con sospecha de malignidad.
-
Informar a los pacientes en tratamiento con hidroclorotiazida acerca de este posible aumento del riesgo tras tratamientos prolongados y advertirles sobre:
- La necesidad de limitar la exposición excesiva al sol o los rayos ultravioleta y de usar fotoprotección solar adecuada.
- La conveniencia de revisar periódicamente la piel y de consultar con un médico en caso de que aparezcan lesiones cutáneas sospechosas (o de que cambien de aspecto las ya existentes).
Para ampliar información:
VARENICLINA Y PÉRDIDA DE CONOCIMIENTO TRANSITORIO
Vareniclina puede producir mareos, somnolencia y pérdida del conocimiento transitoria y, por tanto, puede afectar a la capacidad para conducir y utilizar maquinaria. Deberá indicarse a los pacientes que no conduzcan, manejen maquinaria compleja o realicen actividades potencialmente peligrosas hasta que conozcan si este medicamento afecta a su capacidad para realizar estas actividades.
Para ampliar información:
https://www.aemps.gob.es/informa/boletines-AEMPS/boletinMensual/2018/junio/boletin-junio.htm
APIXABÁN Y EDOXABÁN: INTERACCIÓN FARMACOLÓGICA CON ISRS E ISRSN QUE CONDUCE A UN AUMENTO DEL RIESGO DE SANGRADO
Se ha observado una interacción farmacodinámica entre los anticoagulantes orales directos, apixabán y edoxabán y los antidepresivos inhibidores selectivos de la recaptación de serotonina (ISRS) y los inhibidores selectivos de la recaptación de serotonina y norepinefrina (ISRSN) que conduce a un aumento del riesgo de sangrado.
Es conocido el riesgo de sangrado que presentan los antidepresivos mediante diferentes mecanismos de acción incluyendo la alteración de la agregación plaquetaria, la disminución de los niveles de serotonina en las plaquetas y la reducción del número de plaquetas.
Tras una revisión de la bibliografía publicada, los casos notificados y la plausibilidad biológica, se ha concluido que la administración concomitante de estos fármacos es posible que provoque un efecto aditivo del riesgo de hemorragia. Por lo tanto, la administración conjunta debe emplearse con precaución.
Para ampliar información
https://www.aemps.gob.es/informa/boletines-AEMPS/boletinMensual/2018/mayo/boletin-mayo.htm
QUETIAPINA: REACCIONES ADVERSAS CUTÁNEAS GRAVES Y SOBREDOSIS
Se han notificado reacciones adversas cutáneas graves, incluido el síndrome de Stevens-Johnson, la necrólisis epidérmica tóxica y la erupción medicamentosa con eosinofilia y síntomas sistémicos (DRESS, por sus siglas en inglés Drug Reaction with Eosinophilia and Systemic Symptoms), relacionadas con el tratamiento con quetiapina. Se incluye como nueva reacción adversa asociada al fármaco la erupción medicamentosa con eosinofilia y síntomas sistémicos (DRESS).
Tras una revisión de la bibliografía publicada acerca de la quetiapina y la sobredosis, se ha concluido que en caso de sobredosis con quetiapina de liberación prolongada, se retarda el pico de sedación y el pico del pulso y se prolonga la recuperación, comparado con la sobredosis con quetiapina rápida. Asimismo, también en caso de sobredosis con quetiapina de liberación prolongada, se ha notificado la formación de bezoar gástrico y se recomienda un diagnóstico por imagen apropiado para decidir el tratamiento del paciente.
Para ampliar información
https://www.aemps.gob.es/informa/boletines-AEMPS/boletinMensual/2018/mayo/boletin-mayo.htm
RIVAROXABAN E INTERACCIÓN CON ERITROMICINA, CLARITROMICINA Y FLUCONAZOL
La ya conocida interacción con eritromicina, claritromicina y fluconazol (aumento de la concentración máxima plasmática de la biodisponibilidad de rivaroxaban) es probable que no sea clínicamente relevante en la mayoría de los pacientes, pero puede ser potencialmente significativa en pacientes de alto riesgo. Se han identificado como nuevas reacciones adversas angioedema y edema alérgico, reacciones anafilácticas que incluyen shock anafiláctico, trombocitopenia y síndrome DRESS.
Para ampliar información
https://www.aemps.gob.es/informa/boletines-AEMPS/boletinMensual/2018/mayo/boletin-mayo.htm
RETINOIDES (ACITRECINA, ALITRETINOÍNA, ISOTRETINOÍNA); Actualización de las medidas para evitar la exposición durante el embarazo y de las advertencias sobre efectos neuropsiquiátricos
El motivo de esta revisión ha sido la necesidad de evaluar las posibles mejoras en las medidas de minimización de riesgos relacionadas con la teratogenicidad de los retinoides orales, así como la información actual disponible sobre los efectos neuropsiquiátricos (por ejemplo, depresión, ansiedad, cambios de humor o de comportamiento) que pueden asociarse al uso de retinoides. Para los retinoides tópicos se ha concluido que no tienen asociados estos riesgos, no obstante, se recomienda no utilizarlos durante el embarazo, ni en mujeres que estén planificando un embarazo.
La AEMPS recomienda:
-
Extremar las precauciones para evitar la exposición de retinoides durante el embarazo, cumpliendo estrictamente las condiciones de uso autorizadas:
- Establecer el tratamiento con retinoides orales solo en el caso de que se considere absolutamente necesario.
- Informar sobre el riesgo de malformaciones congénitas y aborto espontáneo asociado a estos medicamentos, así como las medidas preventivas que se deben adoptar durante el tratamiento y posteriormente.
- Asegurar el uso de métodos anticonceptivos por las pacientes con capacidad de gestación, tanto desde el inicio del tratamiento como a lo largo de este y durante un período tras su finalización que depende del medicamento utilizado (hasta 1 mes después para alitretinoína e isotretinoína y 3 años para acitretina).
- Realizar controles periódicos para comprobar la ausencia de embarazo antes, durante y después del tratamiento (sería ideal mensualmente durante el tratamiento y 1 mes después de finalizarlo, en el caso de acitretina se recomienda cada 1 a 3 meses durante los 3 años posteriores).
- En los pacientes en tratamiento, vigilar la posible aparición de síntomas o signos de alteraciones neuropsiquiátricas, como cambios de humor o de comportamiento, en particular en pacientes con antecedentes de depresión. Informar a los pacientes y a sus familiares o cuidadores sobre la posible aparición de estas alteraciones y que acudan al médico en el caso de que estos síntomas apareciesen.
Para ampliar información
OTROS
MEDICAMENTOS PARA LA ENFERMEDAD DE ALZHEIMER. NO REEMBOLSABLES… EN FRANCIA
Desde el 1 de agosto de 2018 los medicamentos para el alzhéimer (donepezilo, galantamina, rivastigmina y memantina) no son financiados por el sistema de salud francés, dado que estos medicamentos tienen una eficacia mínima y transitoria, al tiempo que exponen al paciente a efectos secundarios graves, desproporcionados, e incluso mortales.Es preferible centrarse en ayudar con la organización de la vida cotidiana, el mantenimiento de la actividad, el acompañamiento y la ayuda del entorno.
Esta medida era predecible: a finales de 2016, la Comisión para la Transparencia de la Autoridad de Salud (HAS) llegó a la conclusión de que su beneficio real es insuficiente, por lo que había pedido la exclusión.
Los datos se vuelven cada vez más preocupantes con el tiempo. Los primeros fármacos para la enfermedad de Alzheimer, que aparecieron a mediados de la década de 1990, dieron lugar a una esperanza real. Pero en 1998, después de analizar los datos de evaluación clínica, Prescrire concluyó que su eficacia era solo sintomática y modesta; en cambio, su perfil de eventos adversos parecía aceptable dados los datos conocidos en ese momento.Con el tiempo, los datos sobre eventos adversos graves e incluso fatales se han acumulado, mientras que los datos sobre la efectividad de los síntomas, la dependencia y el resultado de la enfermedad han sido más decepcionantes de lo esperado. Sin mencionar que estos medicamentos están involucrados en muchas interacciones con otros medicamentos.
Para ampliar información
http://www.prescrire.org/fr/3/31/55116/0/NewsDetails.aspx
Bibliografía
- Agencia Europea del Medicamento (EMA). Nuevos medicamentos: Lesinurad. 2018. Disponible en URL: https://ec.europa.eu/health/documents/community-register/2016/20160218134007/anx_134007_es.pdf
- Agencia Española de Medicamentos y Productos Sanitarios. Informe de Posicionamiento Terapéutico de lesinurad (Zurampic®) en el tratamiento de la hiperuricemia. 2018. Disponible en URL: https://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/IPT-lesinurad-Zurampic-hiperuricemia-gota.pdf
- Comité de Evaluación de Nuevos Medicamentos de Euskadi. Informe de evaluación n.º 250/2018. Propiverina. Disponible en URL:
- https://www.osakidetza.euskadi.eus/contenidos/informacion/medicamentos_atencion_primaria/es_def/adjuntos/P/propiverina/propiverina_ficha_es.pdf
- Centro Andaluz de Documentación e Información de Medicamentos (CADIME). Informes de Evaluación de Medicamentos Propiverina. Disponible en URL: http://www.cadime.es/es/fnt.cfm?fid=144Agencia Europea del Medicamento (EMA). Nuevos medicamentos. Prasterona- Intrarosa®. 2018. Disponible en URL: https://www.ema.europa.eu/documents/overview/intrarosa-epar-summary-public_es.pdf
- Agencia Española de Medicamentos y Productos Sanitarios. Prasterona. 2018. Disponible en URL:https://www.aemps.gob.es/cima/publico/detalle.html
- Agencia Española de Medicamentos y Productos Sanitarios. Metamizol y riesgo de agranulocitosis. 2018. Disponible en URL: https://www.aemps.gob.es/informa/notasInformativas/medicamentosUsoHumano/seguridad/2018/NI_MUH_FV-15-2018-metamizol-agranulocitosis.htm
- Shah R. Metamizole (dypirone)-induced agranulocytosis: Does the risk vary according to ethnicity? J Clin Pharm Ther. 2018 Oct 11. Doi: 10.1111/jcpt.12768.
- Agencia Española de Medicamentos y Productos Sanitarios. Quinolonas y Fluoroquinolonas de administración sistémica: nuevas restricciones de uso 2018. Disponible en URL: https://www.aemps.gob.es/informa/notasInformativas/medicamentosUsoHumano/seguridad/2018/NI_MUH_FV-14-2018-quinolonas-fluoroquinolonas.htm
- Agencia Española de Medicamentos y Productos Sanitarios. Hidroclorotiazida: el uso continuo y prolongado en el tiempo podría aumentar el riesgo de cáncer cutáneo no melanocítico. 2018. Disponible en URL:
- https://www.aemps.gob.es/informa/notasInformativas/medicamentosUsoHumano/seguridad/2018/NI_MUH_FV-13-2018-HCTZ.htm
- Pedersen SA, Gaist D, Schmidt SAJ, Hölmich LR, Friis S, Pottegård A. Hydrochlorothiazide use and risk of non-melanoma skin cancer: A nationwide case-control study from Denmark. J Am Acad Dermatol. 2018;78:673-681.e9.
- Pottegård A, Hallas J, Olesen M, Svendsen MT, Habel LA, Friedman GD, Friis S. Hydrochlorothiazide use is strongly associated with risk of lip cancer. J Intern Med. 2017;282(4):322-331.
- Agencia Española de Medicamentos y Productos Sanitarios. Retinoides (acitrecina, alitretinoína, isotretinoína); Actualización de las medidas para evitar la exposición durante el embarazo y de las advertencias sobre efectos neuropsiquiátricos. 2018. Disponible en URL: https://www.aemps.gob.es/informa/notasInformativas/medicamentosUsoHumano/seguridad/2018/NI-MUH_FV_06-Retinoides.htm
- Agencia francesa de seguridad de los medicamentos (ANSM). Médicaments de la maladie d'Alzheimer : enfin non remboursables en France !. Disponible en URL : http://www.prescrire.org/fr/3/31/55116/0/NewsDetails.aspx
Ana Isabel 16-01-19
Muy interesante, claro y conciso. Muchas gracias por esta actualización. Un saludo.