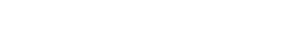






Situación clínica
Paciente de 70 años con antecedentes de hipertensión, accidente vascular isquémico transitorio, ateromatosis de carótidas y enfermedad de Parkinson. Recibe tratamiento con enalapril/tiazida, clopidogrel, atorvastatina, ropinirol, carbidopa-levodopa-entacapona y selegilina. Acude a la consulta por lesiones papulares violáceas de siete días de evolución, con un centro purpúrico palpable y diseminadas por debajo de ambas rodillas hasta el tobillo y el antepié (figura 1). Niega sufrir artromialgias, distermia u otras manifestaciones respiratorias, digestivas y urinarias. Solo presenta sintomatología propia de su enfermedad neurológica. Mantiene buen estado general y la exploración cardiorrespiratoria es normal; no hay signos articulares y un test mediante tira reactiva de orina en la consulta no muestra anomalías. El hemograma resulta normal, con un número de plaquetas conservado, VSG 13, anticuerpos citoplasmáticos antineutrófilos (ANCA) y anticuerpos antinucleares (ANA) negativos. Se le indica al paciente que solo se le debe hacer un seguimiento clínico dado su buen estado general y se comprueba que las lesiones han desaparecido en 10 días.
Puntos clave
La púrpura se define como una lesión eritematosa que no se blanquea a la vitropresión y que se debe a la extravasación de hematíes.
Cualquier dermatosis puede adquirir un carácter purpúrico en su evolución, especialmente las dermatosis de extremidades inferiores, como la dermatitis de estasis.
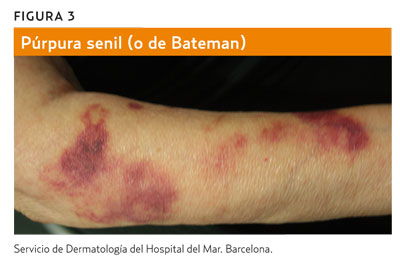
La púrpura de Bateman (senil) es una causa frecuente de equimosis en los antebrazos en personas de edad avanzada.
En Atención Primaria (AP), debe sospecharse una plaquetopenia ante una púrpura no palpable y una vasculitis leucocitoclástica ante una púrpura palpable.
La vasculitis leucocitoclástica suele ser secundaria a fármacos, infecciones o enfermedades internas. Es obligado la detección de la enfermedad renal.
La púrpura de Schönlein-Henoch afecta a pacientes en edades pediátricas después de una infección de vías respiratorias altas; es de aparición aguda con púrpura palpable, artralgias, dolor abdominal y glomerulonefritis.
La evolución de una vasculitis leucocitoclástica es habitualmente favorable y solo suele requerir reposo.
El problema en nuestro medio
La púrpura se define como la presencia de elementos hemorrágicos punteados y diseminados en la piel o mucosas que no se blanquean completamente tras su presión. Pueden observarse lesiones menores a 4 mm (petequias), entre 4 mm y 1 cm y mayores a 1 cm (equimosis). Se trata de una patología común, especialmente en pacientes de edad avanzada, que puede darse por múltiples desencadenantes y constituir un síntoma de una enfermedad sistémica grave. Así, desde la púrpura localizada en una zona corporal hasta la diseminada y generalizada, pasando por la que no muestra sintomatología añadida o la que afecta gravemente el estado general del paciente, la presentación es muy variada y conviene contextualizarla en Atención Primaria (AP).
Aunque no se registran datos bien contrastados de su incidencia en este ámbito, una serie de 20 años identificó 31 casos de Schönlein-Henoch en el adulto frente a 73 en niños1,2. Y en la bibliografía anglosajona se refiere una incidencia anual de entre 20 y 26 casos por 100.000 niños3,4.
Debemos diferenciar la púrpura de la livedo reticularis, que se define como un patrón persistente reticular o moteado, azul, rojo o violáceo, no reversible con calentamiento de la piel, y que puede observarse en el tronco, brazos o piernas en forma de círculos regulares no rotos (livedo reticular regular) o círculos irregulares rotos (livedo racemosa).
¿Cómo podemos orientar una púrpura en una consulta de AP?
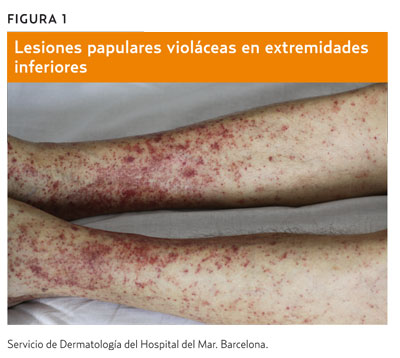
La presentación inicial puede ayudar a clasificarla y a orientar su etiología. Preguntaremos al paciente si donde se halla localizada la púrpura había una lesión previa o agente externo (púrpura secundaria) –como ocurre tardíamente en ciertas picaduras, en algunas celulitis, en la dermatitis de estasis o eccematoide grave (figura 2) o tras traumatismos– o bien la púrpura ha aparecido espontáneamente (púrpura primaria) –en este caso, la hemorragia formaría la lesión íntegramente.
La púrpura primaria es más frecuente y precisa de estudios complementarios. En su abordaje es imprescindible distinguir entre lesiones no palpables y lesiones palpables5 (grado de recomendación C).
¿Cuál es la etiología de las púrpuras no palpables?
Origen cutáneo:
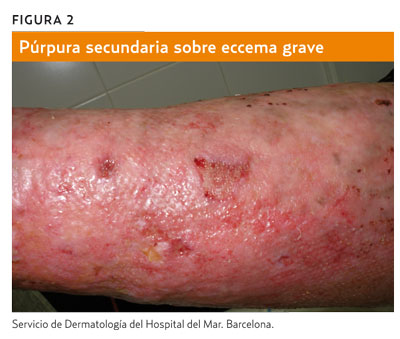
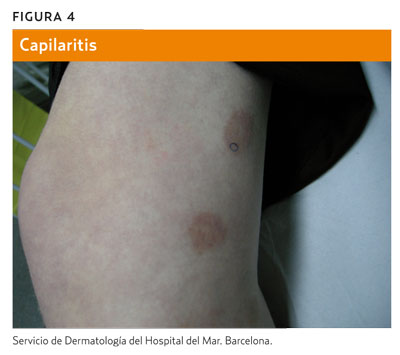
La llamada púrpura de Bateman o púrpura senil (figura 3) consiste en la aparición de equimosis en los antebrazos. Es frecuente en pacientes de edad avanzada. Se debe a la atrofia cutánea del fotoenvejecimiento y se agrava por traumatismos y el uso de antiagregantes, anticoagulantes o esteroides. La capilaritis (figura 4) aparece en forma de petequias dentro de máculas de color marronáceo, de morfología numular, y suele afectar a las extremidades inferiores a causa de una extravasación de hematíes por un proceso inflamatorio perivascular de origen desconocido5.
Origen sistémico:
• Alteraciones de la coagulación: la trombopenia (con cifras menores a 20.000 plaquetas/mm3) incluyendo la púrpura trombocitopénica idiopática (PTI), las alteraciones del funcionamiento de las plaquetas y los defectos de los factores de coagulación.
La PTI es un trastorno adquirido, de mecanismo inmune desconocido, que en adultos se manifiesta de forma crónica con sangrado mucocutáneo (equimosis, petequias, sangrado oral, gastrointestinal o pérdida menstrual abundante) y recuento bajo de plaquetas.
Ciertos fármacos, como el ácido acetilsalicílico y los antiinflamatorios no esteroideos (AINE) pueden inducir un mal funcionamiento de las plaquetas que produzca púrpura.
• La fragilidad capilar subyace en una serie de trastornos poco frecuentes: amiloidosis, Ehlers-Danlos, escorbuto.
• Fenómenos trombóticos en la coagulación intravascular diseminada, la crioglobulinemia, la púrpura trombopénica trombótica (PTT) y la necrosis por dicumarínicos o enfermedad por émbolos de colesterol.
La PTT es una microangiopatía trombopénica trombótica de base genética o adquirida (idiopática). Esta última es más frecuente en mujeres, pacientes con virus de la inmunodeficiencia humana (VIH) o gestantes, o bien está relacionada con medicamentos por la formación de anticuerpos (clopidogrel y ticlopidina). La descripción clásica incluye la presencia de anemia hemolítica microangiopática, trombopenia, insuficiencia renal, sintomatología neurológica y fiebre.
La necrosis cutánea por dicumarínicos se manifiesta con zonas dolorosas de eritema que evolucionan a placas purpúricas y posteriormente a úlceras necróticas. Se observa, sobre todo, en mujeres y en zonas con panículo adiposo abundante (mama, abdomen, glúteos y muslos); aparece entre el tercer y décimo día como consecuencia de un probable déficit en algún factor de coagulación (más frecuentemente de proteína C)6.
La enfermedad por émbolos de colesterol afecta a las extremidades inferiores en pacientes con enfermedad aterosclerótica. Suele observarse tras iniciar anticoagulación o después de una intervención vascular invasiva. Se asocia a otras lesiones cutáneas como livedo, gangrena, cianosis y ulceraciones distales. Debe descartarse una afectación sistémica en forma de insuficiencia renal5.
Otras causas:
La elevación de la presión venosa intravascular durante los fenómenos de Valsalva es causa de púrpura, al igual que fenómenos de presión local, como la que ejerce el manguito del esfingomanómetro, o las correas de las mochilas, entre otras.
¿Cuál es la etiología de las púrpuras palpables?
Debemos tener presente que las púrpuras palpables son siempre de causa sistémica. Según su etiología se clasifican en5,7:
• Vasculitis: representadas por la vasculitis leucocitoclástica o vasculitis de pequeño vaso (inflamación de las vénulas poscapilares), también referida como alérgica o de hipersensibilidad.
Otras vasculitis reconocidas son la enfermedad de Wegener, la vasculitis de Churg-Strauss, la poliangitis microscópica y la crioglobulinemia esencial.
• Causas embólicas: en el contexto de enfermedades infecciosas de gravedad, como la meningococemia aguda, la gonococia diseminada, la fiebre manchada por Rickettsia y la ectima gangrenosa. Suelen acompañarse de fiebre y de afectación grave del estado general. Constituyen una urgencia hospitalaria.
¿Cómo abordamos a un paciente con una púrpura palpable y estado general conservado?
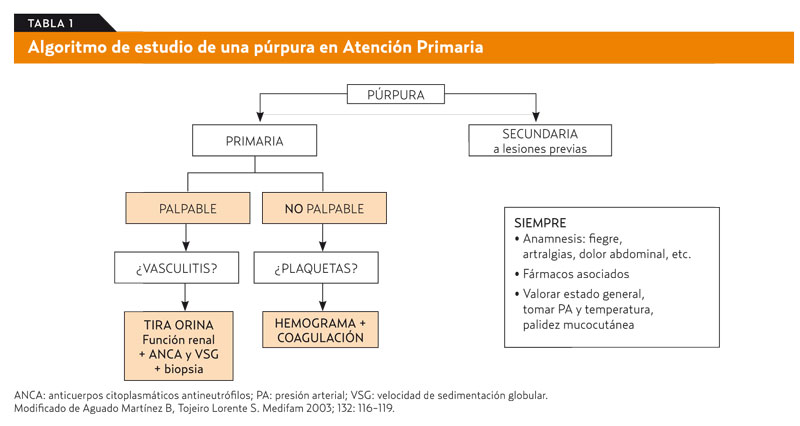
En el ámbito de AP, se insiste en la orientación presentada al comienzo de este artículo: a) si hay una lesión previa sobre la que se ha desarrollado una púrpura, esta será secundaria y perderá entidad y gravedad, yb) si la púrpura es primaria (de novo) y no palpable, debemos pensar en trombopatía o en enfermedades de carácter grave no habituales en el entorno de AP; por el contrario, una púrpura palpable en un paciente con estado general conservado indica que hay una afectación de pequeño vaso en la parte más superficial de la dermis y su origen será casi siempre una vasculitis leucocitoclástica (tabla 1) (grado de recomendación C).

En esta estrategia diagnóstica deberá realizarse una valoración global e íntegra para buscar la etiología más probable y así poder descartar patologías de potencial gravedad, si bien no son tan frecuentes. Por tanto, indagaremos, especialmente, si existen síntomas y signos respiratorios, digestivos, urinarios y articulares, y solicitaremos un hemograma, estudio de orina, serologías de hepatitis y, si es posible, un cribado de enfermedades autoinmunes. La decisión de si efectuar o no una biopsia recae en el especialista en Dermatología (tabla 2) (grado de recomendación C).
En una serie hospitalaria de nuestro entorno, no muy reciente, sobre púrpuras palpables, se describía una etiología muy diversa en los adultos: el 70% de los casos eran vasculitis por hipersensibilidad y púrpuras de Schönlein-Henoch, mientras que el resto se asociaba a vasculitis granulomatosas (13%) y enfermedades del tejido conectivo, enfermedades infecciosas graves y tumores malignos (16,8%)8.
En el caso presentado, la aproximación inicial permite considerar la púrpura como una lesión primaria, pues el paciente negaba de forma tajante haber sufrido lesiones previas en la zona descrita. El carácter palpable de la púrpura y su distribución también llevó a descartar el origen estrictamente cutáneo del proceso, dado que en estos casos las lesiones son típicamente no palpables: exposición solar, esteroides, púrpura senil, capilaritis.
En cuanto a las posibles causas sistémicas de la púrpura, de nuevo el carácter palpable, pero especialmente el número normal de plaquetas, así como el buen estado general del paciente y el cuadro descrito, permiten descartar la trombopatía asociada a fármacos (no concurría un fármaco sospechoso), la enfermedad de las plaquetas o de la coagulación y otras entidades graves (linfoma, leucemia, mieloma, macroglobulinemia, PTT) cuya clínica y expresión analítica no se dan en el paciente. También se puede eliminar como causa los dicumarínicos, pues el paciente no tomaba este medicamento, y la crioglobulinemia asociada a hepatitis C por falta de antecedentes infecciosos en el paciente y por los resultados negativos de la serología.
Únicamente suscita una duda la posible implicación de clopidogrel en el origen del cuadro, pero el efecto secundario más habitual de este fármaco es el sangrado y no la púrpura, y cuando esta aparece lo hace en el contexto de una PTT, es decir, con plaquetopenia y asociada a lesiones no palpables9.
Por tanto, al hallarnos ante una púrpura palpable, consideramos solo dos grupos: la púrpura vasculítica y la púrpura embolígena. Debemos descartar, en primer lugar, el origen embolígeno por la gravedad del contexto en que se producen las lesiones: meningococemia aguda, gonococia diseminada, fiebre manchada por Rickettsia y ectima gangrenosa, lo que excede el cuadro que afectaba al paciente y rarísimamente se incluyen en el diagnóstico diferencial de una púrpura en el entorno de trabajo de la Atención Primaria.
Así, debemos tener en cuenta, por último, la causa vasculítica, entre la que destaca la vasculitis leucocitoclástica o de pequeño vaso.
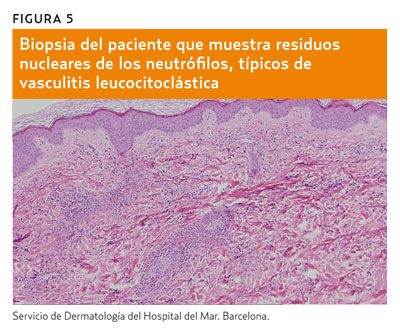
De forma que, tras haber descartado razonablemente las causas de púrpura no palpable y la etiología embolígena de púrpura palpable, nuestra sospecha se dirige, por el contexto clínico, hacia la vasculitis leucocitoclástica o de pequeño vaso, lo que la biopsia confirma (figura 5) y queda ratificado por la benignidad en la evolución de la resolución de las lesiones sin recrudecimiento ni complicaciones posteriores. Nos hallamos, pues, ante una de las vasculitis primarias más frecuentes.
¿Cómo se clasifican las vasculitis primarias?
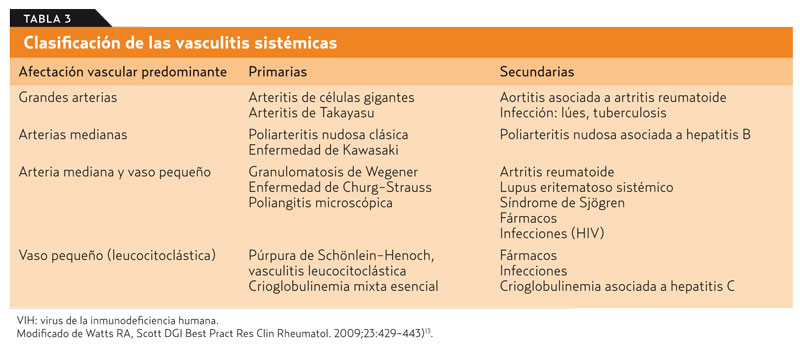
El estudio de las vasculitis sistémicas ha experimentado cambios notables desde 1990 con la introducción de conceptos de gran importancia: el reconocimiento del tamaño dominante de los vasos afectados, la distinción entre primarias y secundarias y la incorporación de marcadores humorales, especialmente los ANCA. Así, en 1990, el American College of Reumathology publicó los criterios de clasificación esenciales10 y, en 1994, la conferencia de Consenso de Chapell Hill consolidó el nombre y la definición para 10 tipos de vasculitis primarias, entre ellas la vasculitis leucocitoclástica (tabla 3). Conviene retener también que entonces quedó establecido que la granulomatosis de Wegener, el síndrome de Churg-Strauss y la poliangitis microscópica son vasculitis asociadas a ANCA11, 12 (grado de recomendación A).
Aun así, la bibliografía todavía muestra lagunas en el conocimiento y clasificación de las vasculitis: la creciente importancia de la hepatitis C en los casos de crioglobulinemia mixta esencial, la infiltración linfocítica en casos de angitis leucocitoclástica o la dificultad de incluir la definición de entidades tan conocidas como el síndrome de Behçet12. También cabe señalar, como característica notable en el estudio de las vasculitis, la dificultad de efectuar el correcto diagnóstico de muchos cuadros. Se insiste en diversos artículos en que los criterios que describen las vasculitis son útiles para la definición y clasificación de casos en epidemiología y ensayos clínicos, pero no deben utilizarse como método de diagnóstico por su bajo valor predictivo positivo12,13.
Una aportación reciente14 propone un algoritmo de estudio de las vasculitis primarias (figura 6) que engloba las seis entidades de pequeño vaso más la poliarteritis nudosa cutánea (PNC). Señalemos que la PNC es una entidad diferente de la poliarteritis nudosa (PAN) en cuanto que parece comprobado que no evoluciona a PAN. La PNC sería una forma crónica recurrente con expresión cutánea, pero de evolución benigna y sin afectación sistémica, según ha evidenciado el seguimiento de los pacientes14,15.
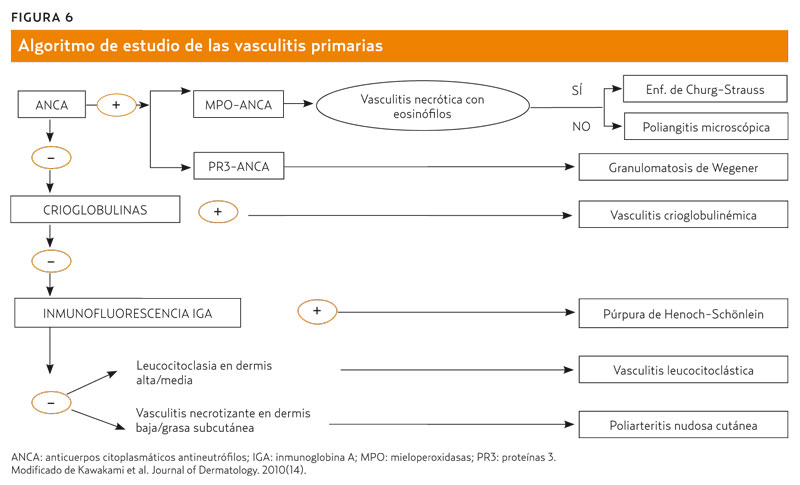
¿Cómo integramos en AP el conocimiento actual sobre las vasculitis?
Para nuestro ámbito de trabajo es importante saber que, aparte de las vasculitis primarias, hay otras patologías de notable entidad, y algunas de ellas de cierta prevalencia, que pueden cursar con lesiones vasculíticas secundarias13. Debemos citar aquí procesos virales inespecíficos, la lúes, la tuberculosis y enfermedades sistémicas como lupus o artritis reumatoide, junto a determinados fármacos: alopurinol, tiazidas, oro, sulfamida, fenitoína y penicilina. La propuesta actual más aceptada para la clasificación de las vasculitis sistémicas en primarias y secundarias se muestra en la tabla 3 (grado de recomendación A)13.
¿Qué debemos saber de la vasculitis leucocitoclástica?
Constituye la causa más común de púrpura palpable. Se atribuye a una reacción de hipersensibilidad anormal frente a un agente infeccioso, un fármaco o antígenos endógenos (inmunoglobulinas), aunque por lo general el antígeno no es identificado. También se habla de «vasculitis predominantemente cutánea». Este grupo de enfermedades son mucho más frecuentes que las vasculitis necrosantes generalizadas (Wegener, poliarteritis nudosa, tromboangitis, etc.).
Se trata de una vasculitis de pequeño vaso que afecta sobre todo a las venas poscapilares. Se caracteriza por un fenómeno denominado «leucocitoclasia», que consiste en la aparición en la pared de los vasos de residuos nucleares de los neutrófilos que participaron en la fase aguda de la reacción inflamatoria. En la fase crónica predominan células mononucleares y a veces eosinófilos. La extravasación de hematíes es consecuencia del daño vascular, que se traduce clínicamente en la lesión purpúrica.
Si bien la púrpura es la manifestación clínica predominante, pueden aparecer otras lesiones cutáneas como máculas, pápulas, vesículas, ampollas, nódulos o úlceras y urticaria crónica y recurrente, y se observan sobre todo en miembros inferiores o en la región sacra en encamados. Suelen acompañarse de escasa afectación sistémica, aunque se describe astenia, artromialgias, febrícula y molestias abdominales. Los estudios de laboratorio son inespecíficos, y es habitual hallar elevación de la velocidad de sedimentación globular (VSG) y ligera leucocitosis con o sin eosinofilia. En ocasiones puede asociarse crioglobulinemia y factor reumatoide positivo16.
La púrpura de Schönlein-Henoch es un subtipo de vasculitis leucocitoclástica que aparece de forma aguda en niños tras una infección de las vías respiratorias superiores. Se caracteriza porque el paciente presenta púrpura en extremidades inferiores y glúteos, así como fiebre, artralgias, dolor abdominal, sangrado digestivo y nefritis. La biopsia cutánea muestra hallazgos de vasculitis por hipersensibilidad con una inmunofluorescencia por inmunoglubulina A (IgA), muy características de la enfermedad. Pero no olvidemos la forma adulta de esta entidad: un cuadro poco frecuente que cursa con púrpura palpable en el 70% de casos, asociada a poliartralgias (menos habitual la artritis) y afectación renal que, si aparece, es insidiosa y obliga a hacer un seguimiento del paciente1. En nuestro caso, no había signos articulares ni urinarios que sostuvieran esta sospecha.
La vasculitis leucocitoclástica se ha asociado a infecciones, fármacos (alopurinol, tiazidas, oro, sulfamida, fenitoína y penicilina), enfermedades del colágeno (lupus eritematoso sistémico, artritis reumatoide y enfermedad de Sjögren), la enfermedad por depósito de crioglobulinas, algunas neoplasias y una constelación de enfermedades en las que la vasculitis es menor y no una manifestación preferente12. En nuestro medio, un estudio antiguo relacionaba la vasculitis leucocitoclástica (citada en el artículo como «de hipersensibilidad») solamente con penicilina y antiinflamatorios no esteroideos17, pero estos datos seguramente están superados por el mayor conocimiento actual.
La mayoría de los casos remiten espontáneamente, por lo que solo debemos recomendar reposo (grado de recomendación A). Es aconsejable informar a los pacientes de posibles reactivaciones antes de su completa remisión. En casos crónicos en los que no pueda tratarse su factor desencadenante o en los que existan manifestaciones sistémicas graves, podemos optar por prescribir tratamientos como corticoides sistémicos, dapsona, colchicine o inmunosupresores18,aunque los escasos ensayos clínicos realizados no ofrecen resultados consistentes (grado de recomendación C)19. El problema surge en el caso de una manifestación cutánea persistente o de la afectación de órganos en los que la respuesta terapéutica no es muy satisfactoria.
Agradecimientos
Expresamos nuestro más sincero agradecimiento a la Dra. Gemma Martín Ezquerra, dermatóloga del Hospital del Mar de Barcelona y de nuestra Área Básica de Salud por sus aportaciones en el artículo y la cesión de las fotografías, además de toda su labor en la consulta ambulatoria.
Lecturas recomendadas
Bolognia JL, Braverman IM. Manifestaciones cutáneas de enfermedades internas. En: Fauci AS, Harrison.a Principios de medicina Interna. 14.ª ed. McGraw-Hill Interamericana de España S.A.U.; 2000. pp. 370-71.
Visión muy general de las lesiones purpúricas y su estudio etiológico.
Watts RA, Scott DGI. Recent developments in the classification and assessment of vasculitis.Best Pract Res Clin Rheumatol. 2009;23: 429-43.
Amplia revisión de la nomenclatura y clasificación de las vasculitis.
Kawakami T. New algorithm (KAWAKAMI algorithm) to diagnose primary cutaneous vasculitis. Journal of Dermatology. 2010;37:113-24.
Descripción y propuesta de un algoritmo de clasificación y diagnóstico de las vasculitis primarias.
Bibliografía
1. García-Porrúa C, Calviño MC, Llorca J, Couselo JM, González-Gay MA. Schönlein-Henoch purpura in children and adults: clinical differences in a defined population. Semin Arthritis Rheum. 2002;32(3):149-56.
2. Domingo Claros A. Problemas hematológicos. En: Martín Zurro A, Cano Pérez JF (eds.). Atención Primaria: Conceptos, organización y práctica clínica. 4.ª ed. Madrid: Harcourt Brace de España; 1999; pp. 1.345-71.
3. Roberts PF, Waller TA, Brinker TM, Riffe IZ, Sayre JW, Bratton RL, MD Schönlein-Henoch Purpura: A Review Article. Southern Medical Journal. 2007;100(8):821-24.
4. Penny K, Fleming M, Kazmierczak D, Thomas A. An epidemiological study of Schönlein-Henoch purpura. Paediatr Nurs. 2010;22 (10):30-35.
5. Bolognia JL, Braverman IM. Manifestaciónes cutáneas de enfermedades internas. En: Fauci AS, Harrison (eds.), Principios de medicina Interna. 14.a ed. McGraw-Hill Interamericana de España; 2000; pp. 370-371.
6. Kurt M, Shorbagi A, Aksu S, Haznedaroglu I, Altundag K, Erkin G. Warfarin-induced skin necrosis and leukocytoclastic vasculitis in a patient with acquired protein C and protein S deficiency. Blood Coagul Fibrinolysis. 2007;18(8):805-06.
7. Aguado Martínez B, Tojeiro Lorente S. Púrpura, todo un reto diagnóstico en la consulta de Atención Primaria: a propósito de un caso. Medifam 2003;13:116-19.
8. Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, García-Fuentes M. Cutaneous vasculitis in children and adults. Associated diseases and etiologic factors in 303 patients. Medicine (Baltimore). 1998;77(6):403.
9. Mangalpally KK, Kleiman NS. The safety of clopidogrel. Expert Opin Drug Saf. 2011;10(1):85-95.
10. Hunder GG, Arend WP, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis. Introduction. Arthritis Rheum. 1990;33:1.065-1.067.
11. Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37:187-92.
12. Saleh A, Stone JH. Classification and diagnostic criteria in systemic vasculitis. Best Pract Res Clin Rheumatol. 2005;19(2):209-21.
13. Watts RA, Scott DGI. Recent developments in the classification and assessment of vasculitis. Best Pract Res Clin Rheumatol. 2009;23:429-43.
14. Kawakami T. New algorithm (KAWAKAMI algorithm) to diagnose primary cutaneous vasculitis. Journal of Dermatology. 2010;37:113-24.
15. Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res. 2009;301:117-21.
16. Koutkia P, Mylonakis E, Rounds S, Erickson A. Leucocytoclastic vasculitis: an update for the clinician. Scand J Rheumatol. 2001;30(6):315-22.
17. García Porrúa C, González-Gay MA, López Lázaro L. Drug associated cutaneous vasculitis in adults in northwestern Spain. Rheumatol. 1999;26(9):1.942-1.944.
18. Martínez-Taboada VM, Blanco R, García-Fuentes M, Rodríguez-Valverde V. Clinical Features and Outcome of 95 Patients with Hypersensitivity Vasculitis. Am J Med. 1997;102(2):186.
19. Sunderkötter C, Bonsmann G, Sindrilaru A, Luger T. Management of leukocytoclastic vasculitis. Journal of Dermatological Treatment. 2005;16(4):193-206.
Marina 27-01-12
molt bé! molt interessant!
Luis Emilio 03-01-12
excelente articulo
Mª Teresa 27-12-11
Me ha gustado mucho la exposición por su claridad y concrecion